Certain nociceptors are described
as “silent” because they do not normally respond to any chemical,
thermal, or tactile stimulus. But when someone suffers an injury and it becomes
inflamed, various molecules associated with the inflammation can lower the activation
threshold of these nociceptors, causing them to “wake up” and begin
producing action potentials. Many nociceptors in the intestines are silent nociceptors.
Both in central and in peripheral
sensitization, there are at least two major mechanisms by which the sensitivity
of neurons is increased. The first comes into play within a few minutes of an
acute injury, but is only temporary. The second emerges more slowly, over a number
of days, but lasts longer. In both cases, the mechanisms are the same as those
described in relation to other types of memorization at the cellular level, in
particular long-term
potentiation.
In the first case, changes occur in proteins that
already exist in the cell membrane and that transduce nociceptive stimuli into
nerve impulses. For example, these proteins may become phosphorylated, that is,
a phosphate group may be added to them by enzymes such as kinases. This phosphorylation
changes the form of the proteins, causing them to become, for example, more permeable
to certain ions. For instance, the AMPA
channel receptor for glutamate is a protein involved in central sensitization.
Phosphorylation increases the likelihood that this receptor’s channel will
open and the time that it will remain open, thus enabling more sodium ions to
enter the neuron and thereby modifying the membrane potential so as to lower the
nociceptor’s overall excitability threshold.
If the nociceptive signal
persists, then the expression of genes or the speed at which their mRNA
is translated into proteins may be increased. If these proteins are new receptors,
then they will be taken to the terminal button of the axon, where they too will
lower the neuron’s excitability threshold and hence increase its sensitivity.
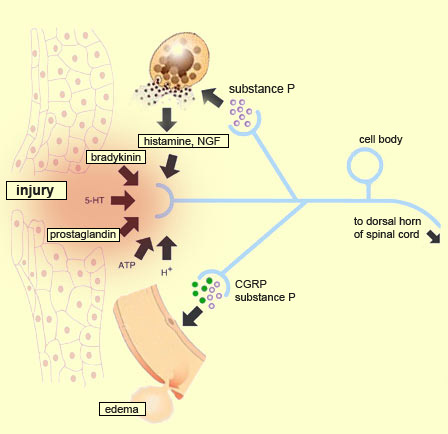
Many different substances contribute
to the sensitization of nociceptors. One of these substances is nerve
growth factor (NGF), which is secreted by fibroblasts (support
cells in connective tissue) and by cells in the epidermis that have been stimulated
by interleukin-1 at the site of an inflammation. The specific receptor for NGF
is the TrkA receptor, which occurs on about 50% of all nociceptors. Activation
of the TrkA receptor results in phosphorylation of the tyrosine residues in its
intracellular portion, as well as phosphorylation of other intracellular molecules
such as the TRPV1 receptors (see diagram to right). This phenomenon might explain
how NGF increases sensitivity to pain caused by heat.
NGF may also contribute
to longer-term sensitization through its ability to modulate the expression of
genes such as those for TRPV1 and P2X3. It is known that the complex that NGF
forms with its TrkA receptor can be endocytosed by vesicles inside the nerve fibre.
Then, by retrograde transport within the axon, the NGF/TrkA complex can ascend
all the way back to the nociceptor’s nucleus, where it activates the synthesis
of numerous peptides, such as substance P and CGRP. It may then also promote the
synthesis of new receptors for the algogenic (pain-producing) peptides secreted
at the site of inflammation, such as bradykinin receptors and vanilloid (VR1)
receptors.
MOLECULES THAT PRODUCE PAIN
Pain is a mechanism that
is essential
to human survival. Pain normally occurs only in the presence of intense stimuli
that are potentially or actually harmful to the body. These stimuli activate high-threshold
nerve fibres called nociceptors
that relay the pain signals via multiple
ascending pathways to the brain.
But unfortunately,
when a tissue injury results in inflammation or in damage to the nervous system
itself, this acute
pain sometimes becomes chronic (in the latter case, it is known as neuropathic
pain). Both inflammation and nerve damage can result in pain that arises spontaneously
in the absence of any apparent peripheral stimuli, or in hypersensitivity to such
stimuli.
The hypersensitivity that occurs after an
injury is not bad in itself. It is even adaptive, because it can promote healing
by making us avoid letting anything come into contact with the injured tissue.
But sometimes this hypersensitivity persists even once the healing is over. In
this case, the resulting pain no longer provides any benefits. Instead, it becomes
chronic pain, a sign of pathological changes in the nervous system.
Understanding
what produces these changes is therefore critically important for treating chronic
pain. We do know that two main mechanisms are involved: central sensitization
and peripheral sensitization. In both cases, regardless of the underlying mechanisms,
the nociceptors become more excitable. In other words, unlike most other human
sensory receptors, which react to repeated stimuli by becoming less sensitive,
when nociceptors are overstimulated, they become more so.
In
the case of central sensitization, this change occurs in the central
nervous system, typically at the synapses between the nociceptors affected
by a severe, persistent injury and the
neurons of the ventral horn of the spinal cord. The injury results in repeated,
high-frequency discharges from these nociceptors, which increases the efficiency
of their synaptic connections to this part of the central nervous system.
In
the injured area, mechanoreceptors that are sensitive to light, tactile stimuli
will then start activating neurons in the spinal cord that normally respond to
nociceptive stimuli only. The gain in the system is thus increased, so that a
piece of clothing simply brushing against the skin can produce a painful sensation.
This phenomenon, known as allodynia, is not the only possible consequence
of central sensitization. The greater sensitivity that may be felt in intact tissue
surrounding the site of an injury is also due to central sensitization and can
lead to chronic
pain.
The
resulting pain is known as “wind-up pain”, which may be either temporary
or permanent, depending on whether the underlying molecular changes are simple
phosphorylations, or more profound changes in the way the genes are expressed
(see sidebar). These changes also reduce the effectiveness of the descending
control mechanisms that use opioids
to alleviate pain.
Such increases in the central nervous
system’s nociceptive response to a given stimulus are not the only mechanism
that can lead to hypersensitivity. A reduction in the excitation threshold of
the nociceptors themselves is another. The term peripheral sensitization
is used to describe what happens when these nerve fibres become more sensitive
than they were before. One classic example is the change in your skin’s
sensitivity that you experience after a sunburn. For example, if you take a shower,
a water temperature that would usually feel hot but comfortable may produce pain
in the sunburned skin.
Here too, a variety of mechanisms,
some faster and others slower (see sidebar), are involved in making the nociceptors’
nerve endings in the affected area more sensitive. When the body suffers an insult
sufficient to cause an injury, the damaged cells release their contents into the
extracellular space. The resulting “molecular soup” then triggers
the secretion of other molecules, in a process known as inflammation. In less
than 15 to 30 seconds, dilation of blood vessels causes the area around the injury
to redden and become warm to the touch. This inflammatory response, which also
causes edema (swelling) and the release of certain chemicals, reaches a peak 5
to 10 minutes later.
The molecules involved in these
local biochemical reactions come from various sources, but their first source
is the damaged cells themselves. For example, this lysis (cell breakdown) usually
releases large numbers of potassium ions, and there is a high correlation
between the concentration of potassium and the degree of pain experienced.
The same is true of the extracellular
concentration of hydrogen ions, which help to activate ion channels
in certain nociceptors directly. This mechanism is responsible, for example, for
the muscle pains associated with the production of adenosine triphosphate (ATP)
under anaerobic conditions, which generates lactic acid during especially heavy
exercise.
Similarly, the ATP from the injured cells contributes to
the depolarization of certain nociceptors by directly activating ATP-dependent
ion channels.
After a tissue is injured, the surrounding tissue
also release substances such as bradykinin (one of the most powerful pain-causing
agents known), histamine, and prostaglandins. By binding to their
specific receptors on the nociceptors’ cell membranes (see box below), these
molecules trigger action potentials in the nociceptive fibres.
Aspirin
and other non-steroidal anti-inflammatories are the reference treatment for these
hyperalgic phenomena, because they inhibit the enzymes involved
in producing prostaglandins.
By a phenomenon known
as the axon reflex, the nociceptors also release substance P into their
peripheral collaterals, that is, into the areas surrounding the injury. This release
of a neurotransmitter in the peripheral nervous system is atypical, because it
proceeds in the opposite direction from usual for a sensory neuron (efferent instead
of afferent). Its effect is to extend and amplify the pain from the injured area.
This release of substance P also causes certain cells, such as mastocytes, to
release histamine themselves, thus further activating the nociceptive fibres in
this broader area. This is why antihistamine creams that block the histamine receptors
can effectively reduce these painful inflammatory reactions.
In
addition to releasing substance P peripherally, the nociceptors also release calcitonin
gene-related peptides (CGRP). Like substance P, CGRP causes vasodilation
both directly, by its effect on smooth muscle tissues, and indirectly, by promoting
the release of histamine by the mastocytes. And by causing edema, this local dilation
of the capillaries will in turn promote the release of bradykinin.
Many
other substances are also involved in peripheral sensitization. One of these is
the serotonin released by the blood platelets, which increases the permeability
of the capillaries and thus contributes to the inflammatory reaction. Another
is nerve
growth factor(NGF), which is important
for the development and survival of the neurons and also plays a role in inflammatory
processes (see sidebar).
Thus we see how complex these
biochemical mechanisms are and how they act in two ways simultaneously: not only
by activating nociceptors directly, but also by lowering their activation threshold,
which is the phenomenon underlying peripheral sensitization.
The nociceptors have numerous transmembrane channels
and receptors that are responsible for transducing chemical,
mechanical, and thermal stimuli. By modifying the conformation of their target
molecules, these stimuli alter the conductance of the cell membrane and hence
induce local currents. If the sum of these local currents is high enough, it will
then trigger action potentials.
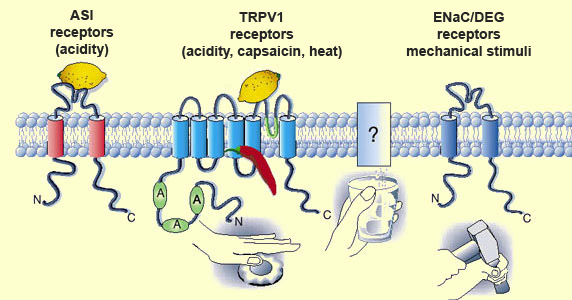
Transient receptor potential (TRP)
channels are sensitive to various kinds of nociceptive stimuli. These channels
are in a sense “generalists” that play a primary role both in nociception
and in other processes of sensory detection. Each TRP is a channel
protein composed of six transmembrane sub-units that allow calcium and sodium
to enter the nociceptor. For example, sub-type TRPV1 (also known as the vanilloid
receptor, VR1) is sensitive not only to capsaicin,
but also to the low pH (acidity) created by extracellular protons, as well as
to heat. Hence it is truly an integrator of chemical and physical stimuli. Researchers
believe that it may also be activated by various kinase proteins, acting through
separate biochemical pathways whose details are far from being fully understood.
In contrast, receptors such as acid-sensing ion channels (ASIC)
are “specialists” that respond to only one type of stimulus: in this
case, extracellular protons, which are either released with the contents of injured
cells or produced by anaerobic respiration—for example, in the form of the
lactic acid that is generated during heavy exercise and makes muscles painful.
Thus, a single type of stimulus can interact with more than one kind of
receptor, as shown by the ability of extracellular protons to activate not only
TRPV1 receptors, but ASIC receptors as well.
The process by which stretching
and mechanical deformation of tissues is translated into pain signals is still
poorly understood. It is thought that proteins in the ENaC/DEG family may
act as mechanical transducers not only in the A
delta nociceptors, but also in the mechanoreceptors. There are also other
candidates among the TRP proteins.
Adapted from Julius and Basbaum, Nature,
2002.
Many other receptors that are modulated by substances given
off by inflammatory reactions can contribute to the generation of pain. For example,
the extracellular ATP produced by inflammation binds to purinergic receptors,
such as P2X3. Activation of the receptors for prostaglandins (PGE2),
bradykinin (B1R and B2R), and substance P (NK1) by their
respective ligands also contributes to the inflammatory reaction.
This
activation often triggers a complex cascade of biochemical reactions. For example,
the NK1 receptor is coupled to a G protein and induces the activation of phospholipase
C, which cleaves its substrate, phosphatidylinositol biphosphate (PIP2), thus
producing inositol triphosphate and diacylglycerol.
An understanding of
the operating mechanisms of these various receptors intimately associated with
nociception will be essential if researchers are to develop new pain medications.
(Existing pain medications, such as opiates
and non-steroidal anti-inflammatories , also act on receptors outside the pain
pathways, thus producing undesirable side effects.)
Chronic use of exogenous opiates
reduces the inhibitory effects that endogenous opioid ligands normally have
on nociceptive pathways. This reduction is explained in part by a decoupling between
the opioid receptors and the G proteins, which short-circuits the cascade of biochemical
reactions that normally follow. The opioid receptors themselves are also desensitized,
while the synthesis of their natural ligands, such as enkephalins, may be diminished.
Lastly, the actual morphology of the neurons is modified by a reduction in the
number of neurofilaments and inhibition of axonal transport.
These changes
cause profound alterations in the neural activity of the pain circuits. The ultimate
outcome is a reduction in the effectiveness of the body’s natural, endorphin-based
analgesic system, along with the phenomena of tolerance
and dependency.
An agonist is a molecule
that acts in the same way as a natural ligand by binding to the same receptor
as that ligand. For example, morphine is an agonist of beta-endorphin, because
it binds to the same mu receptors and produces similar effects. Methadone, a synthetic
compound used to reduce the symptoms of withdrawal
from opiates such as heroin
and morphine, is also an agonist of the mu receptors.
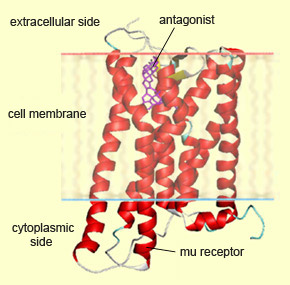
The antagonist
of a molecule is also a molecule that binds to the same receptor as that molecule,
but without producing the same effect. Like a key inserted in the wrong lock,
an antagonist blocks the receptor by occupying its active site, thereby preventing
its natural ligand from binding there. One example of an antagonist that works
this way is naloxone, the best known antagonist for opiates.
For example,
when someone overdoses on heroin, this molecule binds to their opioid receptors
and can thereby reduce their respiration rate to as few as two or three breaths
per minute. But if naloxone is administered intravenously, it quickly competes
with the heroin in the patient’s blood for the chance to bind to these receptors,
and can thereby get the patient’s respiration back up to 15 to 20 breaths
per minute in just a few seconds.
More anecdotally, but also revealingly,
when people who can eat very strong peppers, such as jalapeños, are injected
with opioid antagonists, the enjoyment that these people usually get from these
peppers is quickly replaced by terrible pain, because their natural endorphins
can no longer do their job.
There are also molecules that are regarded
as partial agonists. Partial agonists too occupy the same
kind of receptors as a natural ligand and produce the same effect, but with less
intensity. Increasing the dose of a partial agonist does increase this effect,
but at a certain point, even if the dose continues to be increased, the effect
plateaus. At these very high doses, in the presence of the natural ligand, the
partial agonist behaves somewhat like an antagonist, gradually displacing the
natural ligand from the active sites on the receptors and reducing its effect
accordingly.
Buprenorphin is a partial agonist for mu opioid receptors
and is used like methadone as a substitution treatment for opiate dependency.
The body’s internal opioid
system seems to play a role in psychic
dependency on drugs. For example, it has been shown that mice that have no
mu opiate receptors will not use a device that lets them self-administer alcohol
or cocaine,
whereas normal mice will make extensive use of this device to stimulate themselves.
In an other experiment, rats were injected with morphine
and then placed in a compartment with walls of one colour. The next day, the rats
received a placebo
and were placed in another compartment with walls of a different colour. This
alternating treatment was repeated several times, and the rats thus learned to
associate a particular environment with the pleasurable effects of the morphine.
After this conditioning period, the rats were placed first in one of the
two different compartments, and then in the other, without being injected with
any drug or placebo. When the rats were placed in the environment associated with
the injection of morphine, an increase was observed in the concentration of enkephalins
in the synapses of the nucleus
accumbens in their brains. But when they were placed in the environment associated
with the injection of the placebo, a decrease in this concentration was observed
instead.
This production of enkephalins at a
key site within the brain’s reward circuit therefore seems to be at
least partly involved in the anticipation of a reward. In this connection, it
is worth recalling that dopamine,
a neurotransmitter closely associated with pleasure, is released under the influence
of two types of neuropeptides: on the one hand, cholecystokinins, and on the other,
the enkephalins that bind to mu and delta opioid receptors!
Endorphin receptors are the key
elements in the process by which both endorphins and opioid analgesic medications
suppress pain (for more details, follow the Anesthesics and Analgesics link to
the left). Like most receptors, these endorphin receptors are large proteins
embedded in the neuron’s cell membrane.
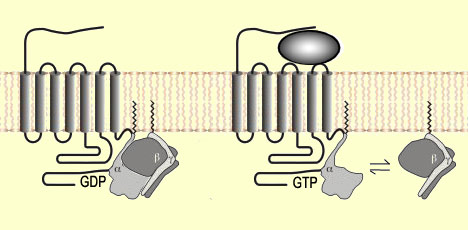
There
are four major families of opioid receptors. All of them consist of proteins that
have 7 transmembrane domains. The portion exposed to the extracellular environment
has a specific site whose form is complementary to that of the opioid substances
that can bind to it. When an opioid binds to such a site, a change occurs in the
form of the receptor, which, on the intracellular (cytoplasmic) side, activates
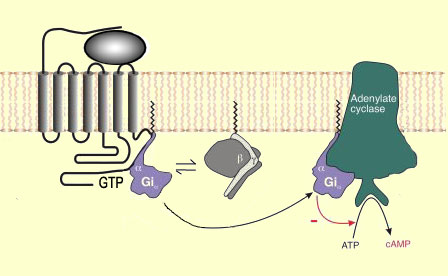
a G protein composed of three sub-units (alpha, beta, and gamma).
When the G protein is activated, the
guanine diphosphate (GDP) molecule that was bound to its alpha sub-unit is replaced
by a molecule of guanine triphosphate (GTP). The GTP in turn causes the alpha-beta-gamma
assembly to break down into an alpha sub-unit and a beta-gamma sub-unit.
Each of these two entities then contributes to
the transduction of the signal. In other words, each of them triggers a cascade
of biochemical reactions inside the cell following a triggering event outside
the cell (the molecules involved in this cascade are often referred to as “second
messengers”). When the event outside the cell is the binding of an endogenous
opioid peptide or an opiate of external origin to an opioid receptor, the effects
of this cascade on the activity of the nerve cell are generally inhibitory.
Among
the inhibitory mechanisms involved, the ones about which the most is known involve
adenylate cyclase, an enzyme that converts ATP
into cyclic AMP. Cyclic AMP is an important second messenger that interacts with
several other proteins, and the reduction of ATP into cyclic AMP is the origin,
for example, of the hyperpolarization
observed in neurons when mu or delta agonists bind to their opioid receptors.
In this case, the cyclic AMP affects potassium channels to produce the decline
in neuronal excitability known as hyperpolarization.
Another
effect of the cascade of second messengers that the G protein initiates inside
the neuron is to reduce the calcium permeability of the voltage-sensitive calcium
channels in the axon’s terminal button (near the synapse). As a result,
in the case of afferent nociceptive C
fibre axons, the amount of neurotransmitters (particularly substance
P and glutamate) released into the synapse is reduced.
The
resulting weakened transmission of the nerve impulse by the presynaptic neuron
has the same effect as the raising of the threshold for triggering action potentials
(hyperpolarization) in the post-synaptic neuron: reduced overall activity in the
ascending pain pathways (see sidebar) and hence reduced perception of pain.
Each
of the major families of opioid receptors also comprises various sub-types that
may be selectively activated by certain ligands. The effects of these ligands
will differ not only according to the particular nature of the receptor sub-type
to which they bind, but also according to the type of neuron on which these receptors
are located.
At least four major families of
opioid receptors are known to exist. They are designated by the Greek letters
mu, delta, and kappa and by the abbreviation ORL1 (where ORL stands for “opioid-receptor-like”).
Delta
opioid receptors were the first type to be described, in the mid-1970s. Enkephalins
are the type of endorphins that have the greatest affinity for delta receptors
and are therefore considered their natural ligands. (Enkephalins can also bind
to mu and kappa receptors, but have less of an affinity for them.)
Researchers initially had difficulty
in characterizing delta receptors, because natural enkephalins have such a short
lifespan. But a bit more has been learned about delta receptors since researchers
succeeded in synthesizing the peptides that are specific to them. For example,
we now know that activation of the delta receptors produces analgesia, though
to a lesser extent than activation of the mu receptors. On the other hand, this
analgesia resulting from activation of the delta receptors also seems to produce
fewer undesirable side effects, and in particular less depressed breathing, constipation,
and tolerance.
Researchers are therefore investigating the treatment of chronic
pain with agonists (see definition in sidebar) that are selective for delta
receptors.
Another interesting characteristic of delta
receptors is their effect in regulating mood. Studies have shown that mutant mice
that did not have the gene for the delta receptor displayed higher levels of anxiety
and depressive behaviour. Here again, agonists specific to delta receptors may
prove invaluable in the treatment of mood disorders.
Delta
receptors are blocked by naloxone, but this opioid receptor antagonist (see definition
in sidebar) has less of an affinity to bind with delta receptors than with mu
receptors.
For mu opioid receptors
, the natural ligand is beta-endorphin,
which was the second opioid peptide to be identified after the enkephalins. Enkephalins
too bind with a high affinity to mu receptors, but dynorphins do not: their affinity
for mu receptors is low.
Mu receptors
are also the preferential binding sites for morphine and the other exogenous opioid
derivatives. However, these morphine-based analgesics produce undesirable side
effects, such as depressed breathing, constipation, and tolerance, mainly by way
of the mu receptor.
Modern
methods of molecular biology have enabled researchers to identify multiple variants
of the gene for the mu receptor. One subtype, mu1, is more associated with the
analgesic effect, and the other, mu2, with the effects on breathing and intestinal
motility.
It has also been proposed that mu receptors
may play a role in bonding between mothers and children, as well as in the reward
circuit. This latter role might be the source of the dependency
behaviours induced by substances such as ethyl alcohol, nicotine, heroin, and
morphine. One piece of evidence for this hypothesis is that mice in which the
mu receptors have been deactivated do not develop such dependencies.
The
distribution of mu receptors in the nervous system corresponds to their effects.
For example, these receptors are present in large numbers in the brainstem centres
that control breathing, as well as presynaptically in the periaqueductal
grey matter and the dorsal horn of the spinal cord, where they contribute
to the descending
inhibition of pain.
This descending control, which
is also involved in the placebo
effect, is blocked by naloxone, a powerful antagonist for mu receptors.
Kappa opioid receptors have
the greatest affinity for dynorphin,
an opioid peptide discovered in the late 1970s. But these receptors too have various
subtypes, and they differ in their affinity for dynorphin. Like the other opioid
receptors, kappa receptors induce analgesia but also cause nausea, as well as
dysphoria and other undesirable psychological effects, which adds to the difficulties
of developing synthetic agonists for this kind of receptor.
Researchers have also found
numerous connections between chronic
stress, its harmful effects on health, and dynorphins. The body’s dynorphin
system, and hence ultimately the kappa receptors that are part of it, are believed
to control certain neural circuits associated with the state of permanent tension
known as stress, thus producing the dysphoric effects associated with it.
More
generally, there is a growing body of data showing that activation of the kappa
receptors produces effects that oppose those arising from the activation of the
mu receptors, such as analgesia, tolerance, reward, and memory. These opposing
effects would also imply that these two types of receptors are located on different
categories of neurons in the circuits where opioid peptides are used.
The most recently discovered
type of opioid receptor is the nociceptin receptor, also
known as ORL1 (where ORL stands for “opioid-receptor-like”).
This receptor has a high affinity for nociceptin (also known as orphanin),
but a very low affinity for the three other major families of endorphins. And
conversely, opioid peptides other than nociceptin scarcely bind at all to ORL1
receptors.
This is a fairly
singular finding, given that the structure of the ORL1 receptor is so similar
to that of the three other classes of opioid receptors. The singularity continues
in terms of the functioning of the ORL1 receptor: depending on the amount of nociceptin
and the site, this receptor may sometimes act as an antagonist to the effects
of opiates, and sometimes as an analgesic itself. The activation of the ORL1 receptor
is also believed to affect levels of dopamine
either directly or indirectly (through the neurotransmitter GABA). Thus, according
to the data currently available, the pharmacology of the ORL1 receptor appears
to be highly complex.
Endorphins and their receptors are
very widely distributed throughout the supraspinal, spinal, and peripheral parts
of the nervous system and are especially numerous in the areas involved in the
descending
control of pain.
Supraspinally, the presence of opioid
receptors in the periaqueductal
grey matter is very well documented and has been shown in autoradiographic
studies. Microinjections of morphine into this structure have been shown to have
a highly analgesic effect, as has electrical stimulation. This effect could be
blocked by naloxone, an opioid-receptor antagonist (see the second sidebar in
this section), etc.
The release of endorphin into the periaqueductal
grey matter can normally be triggered by nociceptive
stimuli from the spinal cord as well as by the connections from many other
structures in the brainstem and higher centres.
Other likely supraspinal
sites for the release of endorphins include the reticular formation, the substantia
nigra, the raphe nuclei, the hypothalamus, the hippocampus, the caudate nucleus,
the amygdala, and the ventral prefrontal cortex.
Spinally, opioid
receptors are located in the axon terminals of C fibres and in the cell bodies
of nociceptive neurons in the surface layers of the dorsal horn of the spinal
cord.
Many interneurons located near the axon terminals of the C
and A delta fibres in these surface layers of the spinal cord release enkephalins
as neurotransmitters. These enkephalins can be released by the activation of the
serotonergic fibres from the reticular formation. The
entryway for the nociceptive signal can thus be blocked either by a reduction
in the substance P or glutamate secreted by the C or A delta fibres, or by a reduction
in the excitability of the nociceptive neurons of the spinal cord. But both are
the result of the binding of enkephalins to their specific receptors.
Peripherally,
opioid receptors have been identified in the terminations of many sensory fibres,
in particular nociceptive C fibres. The three main types of opioid receptors are
produced in the cell bodies of these neurons (located in the spinal ganglia) and
carried by the axons to the peripheral terminations.
The analgesic effect
mediated by these peripheral opioid receptors would appear to be especially strong
in nociceptive fibres that have
already been sensitized by inflammation. Tissue injuries also stimulate
the expression of opioid receptors.
When an opioid medication is administered
either orally or intravenously, it will thus exert its analgesic effects at several
levels. The fact that some immune cells also express opioid receptors indicates
that these effects might be broader still.