|
|
| Funding for this site is provided by readers like you. | |
|
|
|
|
|
|||||
|
|
|||||||
|
|
|
|

| |
Oligomers Help Us Keep Our Memories
| |
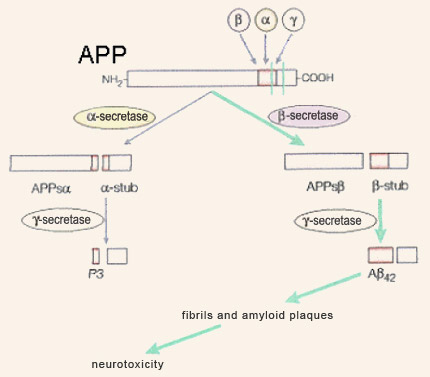
Although amyloid plaques were described by Alois Alzheimer very early in the 20th century, they did not begin to give up their secrets until 1984, when American research pathologist George Glenner and his team characterized their main component: the peptide beta-amyloid. Soon after, in 1992, Hardy and Higgins assigned a central role to beta-amyloid and its precursor, APP (amyloid protein precursor) when they formulated what was to become the most famous hypothesis about the origins of Alzheimer’s-type dementia: the amyloid-cascade hypothesis. According to this hypothesis, the production and subsequent aggregation of beta-amyloid are the ultimate source of the disorders experienced by people with Alzheimer’s. The pathologies associated with the tau protein, as well as other mechanisms proposed to explain Alzheimer’s, are thus regarded as processes downstream from this amyloid cascade. The gene for beta-amyloid’s precursor, APP, is located on chromosome 21 and is expressed in virtually every tissue in the body. This gene, after being transcribed into molecules of messenger RNA which are then spliced in various ways, leads to the production of glycoproteins ranging in length from 695 to 770 amino acids; the form with 695 amino acid predominates in the neurons of the brain. APP is classified as part of a large family of proteins known as transmembrane proteins, because it passes from the inside of the neuron (the cytoplasm) through the cell membrane to the outside. APP’s long N-terminal segment is located outside the neuron, and its short C-terminal segment is located inside. As its full name, amyloid protein precursor, suggests, APP can be cleaved by enzymes to produce other, smaller proteins, including beta-amyloid.
The cleavage of APP by beta-secretases is referred to as its amyloidogenic metabolic pathway, while its cleavage by alpha-secretases is is referred to as its non-amyloidogenic metabolic pathway. The latter may be beneficial, because it may prevent APP from producing beta-amyloid. According to the amyloid-cascade hypothesis, a malfunction in the amyloidogenic pathway results in increased production of the long form of the peptide beta-amyloid—the one with 42 amino acids. Compared with the normal form, which has only 40 amino acids, the form with 42 agglutinates more readily into amyloid plaques. During the mild and moderate stages of Alzheimer’s disease, the form with 42 amino acids predominates in these plaques. It is only later in the progress of this dementia that substantial amounts of the 40-amino-acid form are found there as well. Still according to this hypothesis, the aggregation of 42-amino-acid beta-amyloid first into fibrils (through the juxtaposition of their beta-pleated sheets), and then into fibrillary plaques, poisons the neurons by allowing too much calcium to enter them, causing their death by necrosis or apoptosis. The accompanying inflammatory reaction, causing the cells of the immune system to secrete neurotoxic free radicals, would aggravate this lethal effect. Thus, according to the amyloid hypothesis, the beta-amyloid plaques produce all of the subsequent pathology associated with Alzheimer’s, including neurofibrillary tangles. For many years, this hypothesis represented the dominant paradigm for research on Alzheimer’s, but it has now shown its limitations and become the subject of debate. However, a number of proven facts do continue to support this hypothesis. For example, in 1991, John Hardy’s team discovered that certain cases of the family form of Alzheimer’s disease are caused by APP mutations. In addition, these mutations result in increased production of the long form of beta-amyloid, which is more susceptible to aggregation. These two observations support the idea that the source of the cascade of events that leads to Alzheimer’s can be found in mutations that affect the production of beta-amyloid. However, the most common cases of the family form of Alzheimer’s come from mutations in the gene for presenilin 1 (PS1), located on chromosome 14, and in an analogous gene, called PS2, located on chromosome 1. In 1996, various research teams showed that these mutated PS1 and PS2 proteins also favoured the production of the long form of beta-amyloid, probably by interacting with the catabolism of APP. Also, all indications are that the transmembrane proteins resulting from the genes for presenilin 1 and 2 lie behind the enzymatic activity of the gamma-secretases, which cleave APP at a site that indeed is located somewhere in the middle of the cell membrane. Though not determined by dominant autosomal mutations the way the family form is, the sporadic form of Alzheimer’s is nevertheless influenced by certain genes, among which the gene for apolipoprotein E (ApoE) is the most clearly implicated (see sidebar). In 1993, Judes Poirier’s team showed that the presence of one of the three main forms of the protein ApoE, ApoE4, constitutes a risk factor for both the family form and the sporadic form of Alzheimer’s. Various mechanisms might explain this harmful effect of the ApoE4 variant, including a decrease in the elimination of the beta-amyloid peptide. ApoE also seems to be implicated in forming the amyloid fibrils that agglutinate into plaques. It is also regarded as a co-factor in amyloidogenesis. Other studies, in mice, have shown that ApoE4 reduced the complexity of the dendritic trees of the cortical neurons and the density of the dendritic spines. Other studies also suggest that ApoE4 is less effective than other variants of ApoE in repairing neurons and that it disrupts the induction of long-term potentiation in the hippocampus. Another genetic factor that may predispose individuals to Alzheimer’s and that is attracting increasing attention is the gene for clusterin, also known as apolipoprotein J. This versatile protein has many properties enabling it to protect and repair other proteins that have folded improperly. It may thus bind to the peptide beta-amyloid and prevent it from forming the fibrils that give rise to plaques. Clusterin is also involved in eliminating beta-amyloid peptides and their fibrils. Since the late 1980s, the expression of clusterin has been known to be higher in people with Alzheimer’s, as if their bodies were trying to prevent the development of the amyloid plaques. In the years just prior to 2010, two international consortiums also compared nearly 600 000 genetic markers in over 20 000 people and confirmed the important role that clusterin seems to play in Alzheimer’s. But aside from these data on the production and aggregation of beta-amyloid, there are many observations that are hard to reconcile with the amyloid-cascade hypothesis. For example, even if mice that have the mutations associated with the family form of Alzheimer’s disease produce excessive amounts of the 42-amino-acid form of beta-amyloid (and develop the corresponding amyloid plaques), most of them do not experience significant neuronal losses, show little phosphorylation of the tau protein, and do not have the neurofibrillary tangles that the amyloid hypothesis would predict. And the same inconsistency is observed in certain parts of the human brain, such as the cerebellum, which can have large numbers of amyloid plaques while displaying none of the associated phenomena predicted by the amyloid hypothesis. Significant fuel is added to the controversy surrounding this hypothesis by the poor correlations between the spatial and temporal dynamics of plaque formation and the severity of the observed cognitive deficits. In contrast, cognitive decline in Alzheimer’s is very highly correlated with synaptic loss. This is especially interesting in relation to studies concerning the role that the soluble, non-fibrillar form of beta-amyloid plays in the destruction of synapses. |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
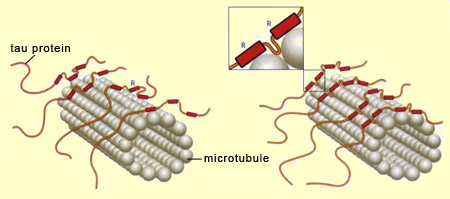
Neurons cannot survive if the nutrients and other substances essential for maintaining their structure cannot travel inside them, and particularly along their axons. These substances are transported through the axons on a network of microtubules which, like the rails on a railroad, ensure that they are carried to the right destinations. When the microtubules are healthy, the neurons can communicate with one another and carry out the normal cognitive functions of the human brain. But when the molecules of tau protein—the “ties” that hold these microtubule “rails” together—come undone, the neurons’ internal transportation networks fail, often resulting in the death of the neurons as well. When this neuronal death occurs on a large scale, it destabilizes the brain’s entire neuronal communication network, resulting in cognitive deficits, and sometimes serious ones such as Alzheimer’s. The term tau pathologies or “tauopathies” designates the entire set of phenomena that result from malfunctions of the tau protein molecules —the “ties” that normally stabilize the microtubules. The tau protein has six variants, all of which are synthesized from the same gene, located on chromosome 17. However, this gene can be read in six different ways, so there are six different types of messenger RNA for this protein, and hence the six variants. The tau protein is distinguished by the repetition of a particular motif, designated R for “repeat”, in its amino-acid sequence. It is this motif that enables the tau protein to bind to the microtubules. Three of the six variants of the tau protein repeat this motif three times, while the other three repeat it four times. Hence these two groups are referred to as 3R variants and 4R variants, respectively. Because 4R tau proteins have one additional binding site, they stabilize the microtubule filaments inside the neurons more effectively than the 3R variants do, thus encouraging longer, more rigid axons. Thus the shape of the neurons can be modulated by the type of tau protein variant expressed.
In addition to the type of tau protein variant expressed (3R or 4R), the stabilization of the microtubules can be modulated even more finely by the degree of phosphorylation of the tau protein molecules. Phosphorylation is a process whereby a phosphate group binds to certain amino acids of a protein. The tau protein has numerous sites where phosphorylation can occur, and the greater the number of phosphate groups that bind to such sites on a tau protein, the less that protein interacts with the microtubules, until ultimately it can become detached from them completely. Tau protein molecules that are thus deactivated by excess phosphorylation then bind to one another to form pairs of spiral filaments. These filaments in turn bind to one another to produce the neurofibrillary tangles that gradually invade the neuron and disrupt its functioning until it is destroyed. Just what causes the
increased phosphorylation of the tau protein remains an open question. One hypothesis
is that free
radicals, due to the presence of beta-amyloid,
are the culprits, and that they act by causing the neuron’s cell membrane
to deteriorate. Calcium ions and beta-amyloid fragments could then penetrate the
neuron more readily and overactivate certain kinase proteins, some of which control
the phosphorylation of tau proteins. |
| |
|
|
|
|
|
|
|
|